Current Status of Clinical Diagnosis and Genetic Analysis of Hereditary Hemorrhagic Telangiectasia in South Korea: Multicenter Case Series and a Systematic Review
Article information
Abstract
Purpose
Hereditary hemorrhagic telangiectasia (HHT), a rare genetic vascular disorder, has been rarely reported in South Korea. We investigated the current prevalence and presenting patterns of genetically confirmed HHT in South Korea.
Materials and Methods
We defined HHT patients as those with proven mutations on known HHT-related genes (ENG, ACVRL1, SMAD4, and GDF2) or those fulfilling 3 or 4 of the Curaçao criteria. A computerized systematic search was performed in PubMed and KoreaMed using the following search term: (“hereditary hemorrhagic telangiectasia” AND “Korea”) OR (“Osler-Weber-Rendu” AND “Korea”). We also collected government health insurance data. HHT genetic testing results were collected from three tertiary hospitals in which the genetic tests were performed. We integrated patient data by analyzing each case to obtain the prevalence and presenting pattern of HHT in South Korea.
Results
We extracted 90 cases from 52 relevant articles from PubMed and KoreaMed. An additional 22 cases were identified from the three Korean tertiary hospitals after excluding seven cases that overlapped with those in the published articles. Finally, 112 HHT patients were identified (41 males and 71 females, aged 4–82 years [mean±standard deviation, 45.3±20.6 years]). The prevalence of HHT in South Korea is about 1 in 500,000, with an almost equal prevalence among men and women. Forty-nine patients underwent genetic testing, of whom 28 had HHT1 (ENG mutation) and 19 had HHT2 (ACVRL1 mutation); the other two patients were negative for ENG, ACVRL1, and SMAD4 mutations.
Conclusion
The prevalence of HHT is underestimated in Korea. The rate of phenotypic presentation seems to be similar to that found worldwide. Korean health insurance coverage is limited to representative genetic analysis to detect ENG and ACVRL1 mutations. Further genetic analyses to detect HHT3, HHT4, and other forms of HHT should be implemented.
INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome, is a rare genetic vascular disorder affecting about 1 or 2 in 10,000 individuals worldwide [1]. It is a systemic disease characterized by recurrent epistaxis, multiple mucocutaneous telangiectasias, and arteriovenous (AV) shunts in various organs. The diagnosis of HHT is based on the Curaçao criteria: 1) spontaneous and recurrent epistaxis; 2) multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; 3) visceral involvement, such as gastrointestinal (GI) telangiectasis, pulmonary, cerebral, or hepatic AV shunts; and 4) a first-degree relative with HHT [2]. The diagnosis of HHT is definite when three criteria are met, possible when two criteria are met, and unlikely when less than two criteria are met.
HHT is inherited via an autosomal dominant pattern. To date, six genes and four related protein molecules have been discovered for HHT and HHT-like diseases (Table 1). The majority of HHT cases are HHT1 ([Online] Mendelian Inheritance in Man, [MIM] #187300) or HHT2 (MIM #600376), with roughly equal proportions of each [3]. More than 1,000 different mutations have been identified as associated with HHT1 and HHT2 [4,5]. Other rare subtypes include Juvenile polyposis-HHT overlap syndrome (JPHT, MIM #175050), HHT5 (MIM #615506), HHT3 (MIM %601101), and HHT4 (MIM %610655). HHT1 is caused by mutations in the ENG gene (chromosomal locus, 9q34.11) and HHT2 is caused by mutations in the activin receptor-like kinase 1 (ACVRL1) gene (chromosomal locus, 12q13.13). JPHT is caused by mutations in the SMAD4 gene.
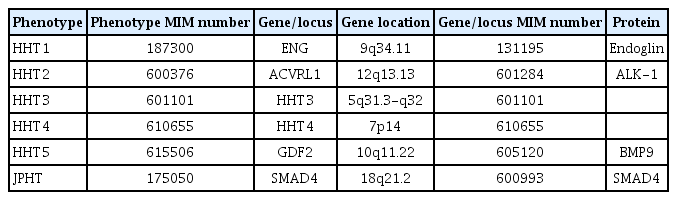
Types of HHT
To our knowledge, there has been no nationwide study focusing on HHT in the South Korean population. This study aimed to review HHT cases in South Korea and compare epidemiologic patterns with global patterns.
MATERIALS AND METHODS
Article search and case retrieval
For the purposes of this study, we defined HHT patients as those with proven mutations on known HHT-related genes (ENG, ACVRL1, SMAD4, and GDF2) or those fulfilling three or four of the Curaçao criteria. The inclusion criteria for relevant articles were 1) case report or case series containing at least one HHT patient and 2) articles with first and corresponding authors of Korean nationality from Korean institutions. Exclusion criteria included 1) articles that did not list the authors’ affiliations and 2) articles without sufficient patient information. A computerized systematic search was performed in PubMed using following search term: (“hereditary hemorrhagic telangiectasia” AND “Korea”) OR (“Osler-Weber-Rendu” AND “Korea”). The literature search has not limited any filters including language. Additional searches were performed on the KoreaMed online database using the search term: (“hereditary hemorrhagic telangiectasia”) OR (“Osler-Weber-Rendu”). The search was updated until December 2018. A bibliographical search was performed to retrieve relevant articles not found on PubMed and KoreaMed (Fig. 1).
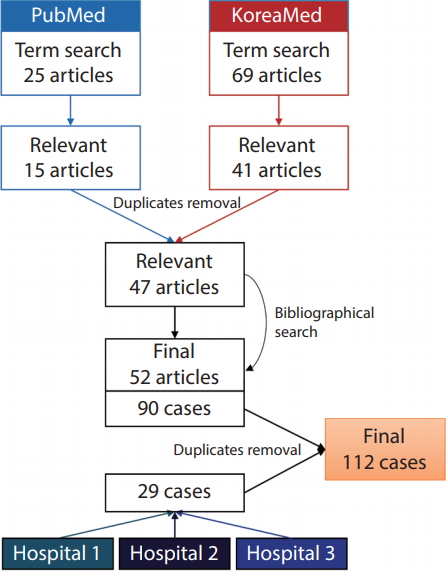
Flow diagram of article search and case retrieval.
The electronic medical records (EMRs) of three tertiary hospitals in South Korea (Asan Medical Center, Seoul, Korea; Samsung Medical Center, Seoul, Korea; Severance Hospital, Seoul, Korea) were searched for HHT genetic testing results (those positive for ENG, ACVRL1, SMAD4, or GDF2) as well as the diagnostic terms “hereditary hemorrhagic telangiectasia” and “Osler-Weber-Rendu.” We analyzed government health insurance data using disease codes and found that there were about 100 HHT patients registered annually in Korea between 2010 and 2017. The data were retrieved using the disease code of I780 (HHT) and I280 (pulmonary arteriovenous fistula) from http://opendata.hira.or.kr/.
Case analysis
Major clinical manifestations, including the presence of epistaxis, GI bleeding, other mucocutaneous telangiectasias, pulmonary arteriovenous malformations (AVMs), hepatic AV shunts, and central nervous system (CNS) AV shunts were recorded for each case (Fig. 2). Other remarkable clinical manifestations that may have been HHT-associated were also recorded. Patient information—such as sex, age, results of genetic tests, and the presence of a family history of HHT—was also recorded (Fig. 3). The cases were thoroughly inspected for duplicates, and duplicate cases were removed. Missing data for each case were considered negative, under the postulation that the patient would not undergo specific evaluation if the corresponding symptom was absent. The number of missing data points corresponding to each manifestation was mentioned. The percentages of each major clinical manifestation, as well as other remarkable clinical manifestations, were calculated for all HHT patients, patients with HHT1, and patients with HHT2.
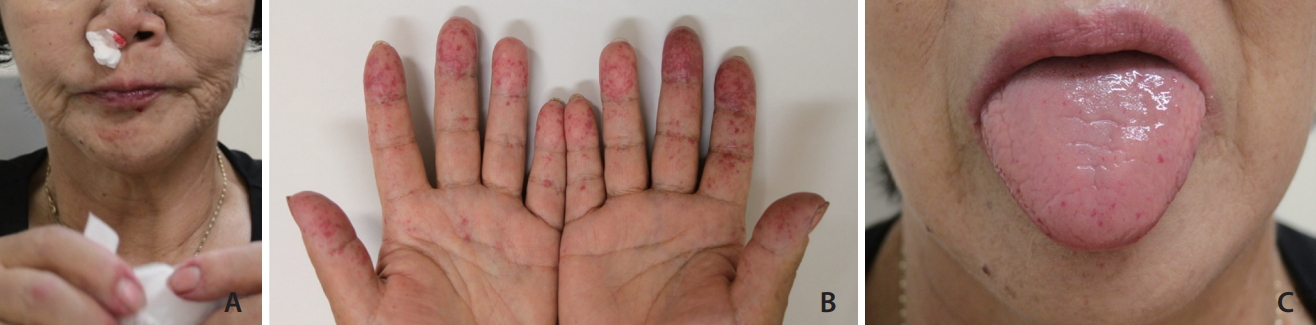
A 60-year-old female with clinically confirmed human hereditary telangiectasia presented with repeated epistaxis that required embolization (A). Note mucocutaneous telangiectasias in hands (B) and tongue (C).

A family tree of a 7-yearold boy (proband, arrow) who presented with multiple brain arteriovenous fistulae. Positive sign (+) indicates the presence of the ENG c.808C>T (p.Gln270Ter) hetero variant; negative sign (-) indicates its absence. DM, diabetes mellitus; AVF, arteriovenous fistula; AVM, arteriovenous malformation; ICH, intracranial hemorrhage.
RESULTS
A total of 25 articles were found on PubMed, 15 of which were deemed to be relevant. A total of 69 articles were found on KoreaMed, 41 of which were relevant. After excluding the duplicates, 47 of these aforementioned articles were retained for the analysis. An additional six relevant articles were found after reviewing the bibliographies of these 47 articles. We extracted a total of 90 cases from 52 articles (Supplementary Material). An additional 29 cases were identified from the three Korean tertiary hospitals; these cases were thoroughly inspected for duplicates, and seven duplicate cases were identified and excluded (Fig. 1). Finally, 112 HHT patients were analyzed (41 males and 71 females aged 4–82 years [mean±standard deviation, 45.3±20.6 years]).
The HHT cases with major clinical manifestations are listed in Table 2. Other remarkable clinical manifestations included stroke (n=5), intracranial hemorrhage (n=3), cerebral or cerebellar abscess (n=4), intracranial arterial aneurysm (n=2), T1-high signal intensity of basal ganglia on brain magnetic resonance image (MRI) (n=3), cerebral cortical dysplasia (n=1), and patent foramen ovale (n=2). Forty-nine patients underwent genetic testing, of whom 28 had HHT1 (ENG mutation) and 19 had HHT2 (ACVRL1 mutation); the other two patients were negative for ENG, ACVRL1, and SMAD4 mutations. The HHT1 and HHT2 cases with major clinical manifestations are listed in Table 3. The mutations were compared with the ENG and ACVRL1 database from the ARUP Laboratories at the University of Utah [4,5], and this identified five novel ENG mutations and four novel ACVRL1 mutations. All identified ENG and ACVRL1 mutations and their corresponding major clinical manifestations are listed in Tables 4 and 5.

Major clinical manifestations of HHT patients in Korea

Major clinical manifestations of HHT1 and HHT2 patients in Korea
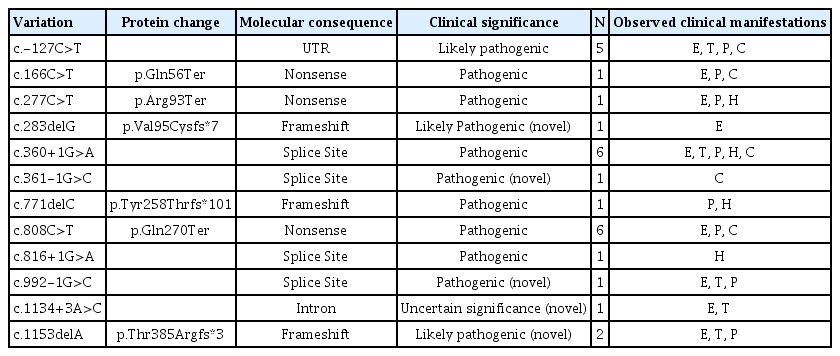
List of ENG mutations identified in Korea

List of ACVRL1 mutations identified in Korea
Health insurance data of Korean government found that there were about 100 (99–133) HHT patients registered annually in Korea between 2010 and 2017. The annual number of new patients was not identified. The outpatient-to-inpatient ratio was about 42:58 during this time. According to health insurance data, the prevalence of HHT in South Korea is about 1 in 500,000, with an almost equal prevalence among men and women [6]. This is far lower than the worldwide prevalence, which may indicate an underestimation of the disease entity in Korea. HHT types other than HHT1 and HHT2 were not identified because South Korean health insurance policies only cover genetic testing for ENG and ACVRL1.
DISCUSSION
Clinical manifestation of HHT
Recurrent epistaxis is usually the earliest clinical sign of HHT. The mean age of onset of epistaxis is 12 years, and it occurs in more than 90% of HHT patients [7]. The degree, interval, and duration of epistaxis vary by patient. The main source of epistaxis is telangiectasis of the nasal mucosa. Recurrent GI bleeding is observed in 20% to 30% of patients with HHT. The main source of GI bleeding is telangiectasis of the GI mucosa. Upper GI telangiectasias are more common than lower GI telangiectasias [8]. Both epistaxis and GI bleeding can result in iron deficiency anemia and may require blood transfusion in severe cases [9]. Mucocutaneous telangiectasis involving the skin and oral mucosa occurs in more than half of HHT patients, usually in later life [10]. The fingertips are the most common location of cutaneous telangiectasis.
Dilatation and malformation of larger visceral vessels can occur in HHT and can manifest as AV shunts. Pulmonary AVM is the most common type of AV shunt, occurring in almost half of HHT patients. AVMs are clinically important in that they are anatomical right-to-left shunts, and they may cause hypoxemia, paradoxical embolic cerebral infarctions, or cerebral abscesses [11]. Hepatic AV shunts are identified in 30% to 60% of HHT patients. Complications of hepatic AV shunts include hepatic encephalopathy due to portovenous shunts; portal hypertension, heart failure, and pulmonary hypertension due to large left-to-right shunts and high cardiac output; and pseudocirrhosis or cirrhosis of the liver [12]. Pulmonary AVMs and CNS AV shunts are more common in HHT1 than HHT2, whereas hepatic AV shunts are more common in HHT2 [1].
Our case analysis showed overall lower incidence of clinical manifestations, compared with worldwide incidence. This may be due to considerable missing data due to the nature of this study, and thus is the limitation of our study. However, the ratios of incidences of clinical manifestations between HHT1 and HHT2 were similar to that of worldwide. The rate of epistaxis was similar in HHT1 and HHT2. There was no case of GI bleeding in HHT1 and rate of hepatic AV shunt was lower than that of HHT2. On the other hands, there was no case of cerebral AV shunt in HHT2 and the rate of pulmonary AVM was lower than that of HHT1.
Neurological manifestations of HHT
The neurologic manifestations of HHT include embolic stroke, abscess formation, migraine, hemorrhagic stroke, and seizures [13]. The most frequent complications are stroke and cerebral abscess, mainly caused by paradoxical embolism [14]. Pulmonary AVMs—which are reported in 20% to 30% of HHT patients—are the most frequent manifestations of the disease and are responsible for a right-to-left shunting leading to paradoxical embolism, causing stroke or cerebral abscess formation.
CNS AV shunting is noted in 10% of HHT patients. Unlike pulmonary AVMs, CNS AV shunts also occur in infants and young adults with a tendency toward multiplicity, small size, and cortical location. CNS AV shunts include cerebral AVMs, cerebral micro-AVMs, high-flow arteriovenous fistulas (AVFs), and telangiectasias [15]. They are usually silent but can present with seizures, ischemia, or hemorrhage. These complications are associated with a higher risk of subsequent hemorrhage, as is the case among patients with sporadic brain AVMs [16]. High-flow AVFs are most commonly found in children.
Developmental venous anomalies and cerebral aneurysms can also be found in HHT patients [17]. Malformations of cortical developments (MCDs), mostly polymicrogyria, are sometimes observed in association with HHT [18]. The exact pathogenetic mechanism for MCDs is not well established; however, it is postulated that decreased expression of ENG may result in focal hypersprouting angiogenesis during corticogenesis, leading to MCD [19]. All cases reported to date have been associated with HHT1, and the lesions are characteristically unilateral, focal, and correlate with arterial regions exposed to the lowest fluid shear stress in utero [19].
Genetic biology in HHT
HHT is characterized by sporadic diameter deviations in smaller and larger vessels, and thus frequent AVMs. AVMs lead to the formation of direct connections between feeding arterioles and the draining veins that shunt blood flow and effectively bypass the capillary network. Between 85% and 90% of HHT genetic drivers are linked to impaired activity of the bone morphogenetic protein (BMP) pathway, and these include heterozygous loss-of-function mutations in endoglin (ENG/CD105), in activin A receptor-like kinase 1 (ACVRL1/ALK1), and in cytosolic sterile alpha motif domain-containing 4A (SMAD4) [20].
A candidate gene approach identified mutations in the transforming growth factor (TGF)-β coreceptor gene ENG in patients with HHT type 1, whereas mutations in the TGF-β type I receptor ALK1 (ACVRL1) cause HHT type 2 [21]. Both genes are predominantly expressed in endothelial cells and have been shown to be regulated by hemodynamic forces in mice and zebrafish. Mutations in ENG, which encodes an auxiliary receptor in the TGF-β superfamily signaling pathway, are responsible for HHT type 1, characterized in part by blood vessel enlargement. Mutations in components of the TGF-β superfamily of signaling molecules disrupt this pattern of hierarchically ordered blood vessel trees and cause AVMs. ENG is a TGF-β coreceptor that enhances signaling through the type I receptor ALK1.
In contrast to what was reported in early studies, endothelial cell proliferation is regarded as a secondary event, driving further enlargement but not the initiation of AVM. A recent report highlighted clonal endothelial proliferation as a major effect of deficient BMP signaling downstream of ALK1-ENG signaling [20]. BMPs 9 and 10 have been known as the primary ligands for ALK1/ENG. Importantly, ENG potentiates ALK1 pathway activation downstream of hemodynamic forces [22]. Inducible homozygous endothelial-specific deletion of ALK1 or ENG during development consistently drives AVMs; however, additional triggers in the adult—such as inflammation, wounding, or VEGF overexpression—are required for AVMs to develop [23]. A full understanding of the mechanisms of vessel diameter control and AVM formation will require insights into quantitative relationships between cell numbers, their movement, their relative shape changes and mutual dependencies on flow, and the gene dose and signaling dose responses in this adaptive system [20].
Screening of family members with HHT
The life expectancy of unscreened and untreated HHT patients is lower than that of people without HHT with an earlier median age of death ranging from 3 to 7 years [13,24]. HHT guidelines recommend screening children with suspected or diagnosed HHT for pulmonary AVMs and cerebral AVMs. The choice of screening tests for pulmonary AVMs should be decided on a case-by-case basis. The neurologic complications of HHT attributed to these AVMs include embolic stroke, cerebral abscess formation, migraine, hemorrhagic stroke, and seizures [13]. Screening for cerebral AVMs in children is recommended by HHT guidelines, and it is performed in North American HHT centers; however, in some other countries, screening for cerebral AVMs is postponed until adulthood [24,25]. Unenhanced magnetic resonance imaging is recommended to detect cerebral AVMs as early as possible or at the time of diagnosis, preferably in the first 6 months of life [26]. If no cerebral AVMs are detected by this scan in adulthood, no further screening for cerebral AVMs is recommended. Screening for the presence of HHT and of pulmonary and cerebral AVMs, combined with treatment if indicated, will prevent severe complications and will result in a similar life expectancy compared to that of the general population [24].
Limitations of this study
There were several limitations in this study. First, as mentioned above, there might be some missing data in collecting patients with HHT. However, the methodology to find out specific presenting patterns according to the subtype of HHT is limited due to rarity of the disease and diversity of the symptoms. Second, genetic studies were not performed in more than half of the cases. Therefore, analysis of the subtype of HHT may be incomplete for those who did not undergo a genetic study. Third, although we eliminated the overlaps of cases, there is still a possibility of duplicated cases. A large-scale nationwide registry or cross-sectional study is warranted in the future.
CONCLUSION
The prevalence of HHT is underestimated in Korea. The rate of phenotypic presentation seems to be similar to that found worldwide. Although pulmonary AVF formation was the most common vascular manifestation, hepatic AVM seems to overlap with liver cirrhosis and hepatocellular carcinoma, which are relatively common diseases in Korea. Cerebral AVMs were also found, particularly in association with HHT1. However, spinal AVMs have not been reported in the Korean literature. Polymicrogyria is rare but has been recognized in Korean patients with HHT. In addition to clinical diagnoses made using the Curaçao criteria, gene analysis can identify HHT in patients and their families. Korean health insurance coverage is limited to representative genetic analysis to detect ENG and ACVRL1 mutations. Further genetic analysis to detect HHT3, HHT4, and other forms of HHT should be implemented for patients who are clinically diagnosed as having HHT.
SUPPLEMENTARY MATERIALS
Supplementary materials related to this article can be found online at https://doi.org/10.5469/neuroint.2019.00150.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2018R1A2B6003143).